


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
9 Minutes
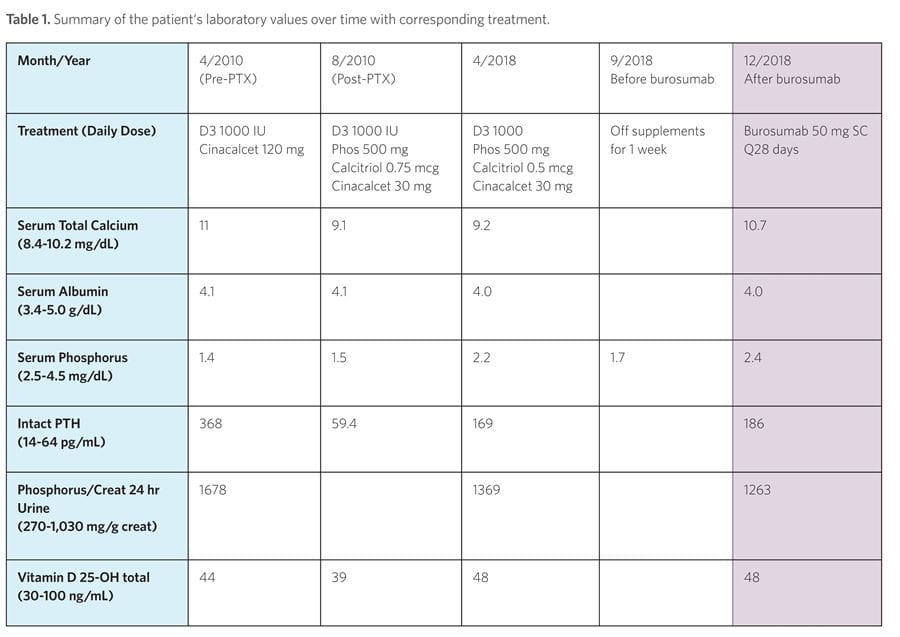
The patient began experiencing progressive and debilitating knee pain secondary to degenerative joint disease related to her bowing deformities. She eventually underwent bilateral knee replacements in her late 40s (Figure 1). She also experienced declining bone mineral density with loss of height but without fragility fractures. In addition, she developed progressive hearing loss, as well as recurrent dental abscesses since age 10, necessitating extensive dental work.
Notably, the patient had been evaluated and followed at many medical centers over her adult life. In 2010, genetic testing was done at another institution and no mutation of the PHEX gene was identified, but the patient’s FGF23 was high at 659 pg/mL (reference range: 10-50 pg/mL). Her family history was negative for any relatives with documented XLH. However, one of her brothers had mild bowing deformities of the legs, and two of his children had recurrent dental caries.
On physical exam, she had short stature (143 cm) with a disproportionally short lower segment compared to the upper segment with visible bowing deformities of her femurs. Her face was asymmetric and she had multiple fillings and healed implants on oral exam. Her spine was notable for moderate to severe scoliosis with loss of lumbar lordosis.
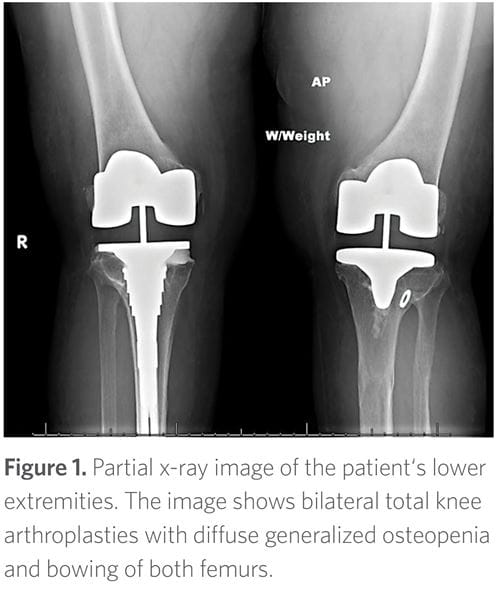
In September 2018, her phosphorus and calcitriol supplements were discontinued, as well as her cinacalcet in preparation for beginning burosumab/KRN23 (Crysvita), the first FDA-approved treatment for X-linked hypophosphatemia. Her serum phosphorus level in the absence of the above supplements and prior to burosumab therapy was 1.7. On September 24, 2018, she received her first monthly burosumab injection of 50 mg (1 mg/kg). Her serum phosphorus increased and has remained in the 2.2-2.7 range without supplements since starting the drug (Table 1). Overall, the patient states her quality of life has dramatically improved since starting burosumab.
X-linked hypophosphatemia (XLH) (also known as X-linked hypophosphatemic rickets) accounts for ~80 percent of familial cases of hypophosphatemia.1 The disease was first described in 1937 by Albright, Butler, and Bloomberg as a rare case of vitamin D-resistant rickets requiring more vitamin D than the amount ordinarily effective for prevention and cure of rickets.2
In 1995, the causative gene for XLH was identified on chromosome Xp22.1 and named PHEX (Phosphate regulating endopeptidase on the X chromosome).4 This gene encodes a cell surface-bound protein-cleaving enzyme. Its expression is enriched in bone and teeth.4 Loss of function mutations in PHEX lead to an increase in fibroblast growth factor-23 (FGF23), a hormone produced predominantly in bone.4 High free FGF23 in plasma causes hypo-phosphatemia and hyperphosphaturia by decreasing renal phosphate reabsorption through downregulation of the phosphorus transporters NaPi-IIa and NaPi-IIc at the apical membrane of proximal renal tubular cells. High FGF23 also leads to inhibition of 1alpha-hydroxylase, resulting in decreased 1,25 vitamin D levels.5
Low serum phosphorus levels are usually present at birth but often remain undetected. For this reason, affected individuals may not present until weight-bearing age, when abnormal mineralization (rickets) leads to bowing deformities of the legs and progressive departure from normal growth rates.8 Progressive enthesopathy, craniosynostosis, bone pain, insufficiency fractures, and dental abscesses also are common findings, particularly in adults.8
Younger siblings of affected patients should be screened to diagnose the disorder before complications develop. Screening can be accomplished by measuring fasting serum phosphorus, as well as renal phosphorus excretion. Laboratory abnormalities at the time of diagnosis often include the following: low serum phosphorus with high urinary phosphorus excretion; elevated FGF23; normal serum calcium, 25-hydroxy vitamin D and PTH levels; elevated alkaline phosphatase; and absence of the normally expected rise in the concentration of 1,25-dihydroxy vitamin D in response to the low serum phosphate.7 In addition, the diagnosis can often be confirmed with genetic testing for PHEX gene mutations, which account for up to 87 percent of cases.13 It is important to note, however, that a PHEX gene mutation cannot be identified in all patients. Novel mutations in the PHEX gene are still being identified, including 14 new mutations that were reported in a cohort of 59 adults with XLH in 2017.14 This could explain the negative genetic testing in this patient in 2010 and can be confirmed with whole genome sequencing.
Most children with XLH are treated with oral phosphorus supplements and 1,25 vitamin D from initial diagnosis until growth is complete.7 Initiation of treatment in early infancy results in improved patient outcomes but does not completely normalize skeletal development or fully correct the metabolic abnormalities.9 The response to treatment is often variable.
Osteomalacia and spontaneous insufficiency fractures should trigger treatment in adults. Treatment with calcitriol and phosphate supplements decreases bone pain, increases serum phosphorus, and reduces osteomalacia as quantified by bone biopsies.10 Long-term complications with standard therapy in both children and adults include hyperphosphaturia with nephrocalcinosis and secondary hyperparathyroidism.7 Treatment with calcitriol and phosphorus requires frequent monitoring and dose adjustments to minimize complications.
As exemplified in this case, a transformative therapy for patients suffering from XLH was approved by the FDA in April 2018. Burosumab/KRN23 (Crysvita), a human monoclonal antibody to FGF23, is the first therapy for X-linked hypophosphatemia that specifically targets the underlying causative mechanism. Studies in adults have demonstrated that subcutaneous administration of KRN23 every four weeks for 16 months increases serum phosphorus levels to the low normal range in 94 percent of subjects and increases 1,25(OH)2D from baseline in all subjects, consistent with inhibition of FGF23 activity.11 In children, treatment with burosumab administered once every two weeks provides a sustained increase in serum phosphorus levels to normal or near-normal levels.12 In addition, studies in both adults and children have revealed improved healing of fractures and pseudo-fractures, as well as improvement in osteomalacia on bone biopsy. Adults also report substantially improved quality of life. Current recommendations for dosing are 0.8 mg/kg every 14 days for children and adolescents and 1 mg/kg every 28 days for adults. The maximum dose is 90 mg for children and adults. All phosphorus and 1,25 vitamin D supplements must be discontinued one week before beginning burosumab with close monitoring for hyperphosphatemia during treatment.
