


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
7 Minutes
This article was written by Tariq Latif, MD, and Frederick DeRubertis, MD.
A 57-year-old Caucasian male presented to his primary care physician (PCP) with persistent lightheadedness. His lightheadedness had been going on for six months and was positional in nature, with his lightheadedness becoming more prominent when he extended his neck. In addition, he also had associated left-sided neck fullness, dry cough, and palpitations. Patient denied any weight loss, fevers, headaches, vision changes, difficulty swallowing, or changes in his voice.
The patient’s past medical history included hypertension (diagnosed five years prior), primary hypothyroidism, eosinophilic esophagitis, and gastroesophageal reflux. He did not have any prior head and neck surgeries or radiation. The patient denied any known family history of endocrine syndromes or neuroendocrine tumors. Likewise, he denied prior tobacco and alcohol use. His blood pressure was well controlled on losartan 25 mg daily alone. Patient was not on any anti-depressants or anxiolytic medication. On physical exam, he was found to have a blood pressure of 134/82, and heart rate was 102. Additionally, he was found to have significant orthostasis on orthostatic blood pressure check. His neck examination was significant for a left-sided palpable lesion with no obvious locoregional lymphadenopathy. Neurological exam was non-revealing, and cranial nerve function was intact.
Given the patient’s symptoms, his PCP ordered a neck ultrasound, which showed a left cervical level II heterogenous hypoechoic vascular mass located within and expanding the carotid sheath with the superficial visible surface measuring 2.9 x 1.2 x 2.0 cm. This prompted a CT of the head and neck with IV contrast, which showed a large mass extending from the level of the left carotid bifurcation superiorly to the left skull base with encasement of the left internal carotid artery with the mass measuring 4.3 x 2.0 x 2.1 cm, with suspicion for paraganglioma; CT also showed several enlarged lymph nodes on the left with the largest being 1.1 cm in the level IIB region (see image 1). At this time, the patient was referred to endocrinology at a satellite UPMC facility as well as ENT.
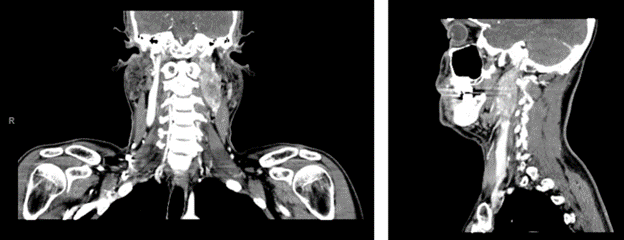
Image 1. CT scan with contrast of head and neck showing the left carotid sheath paraganglioma.
The patient underwent biochemical testing, which showed elevated serum normetanephrines of 252 pg/mL and elevated serum metanephrines of 155 pg/mL, both done by LC/MS/MS. 24-hour urinary fractionated normetanephrines and metanephrines were also elevated to three times the upper limit of normal. A 24-hour urine dopamine level was normal. Patient then underwent a dotatate PET/CT scan, which redemonstrated the same cervical carotid sheath mass with associated lymphadenopathy — all of which were strongly dotatate positive/avid (see image 2). After this, the patient was started on alpha blockade with doxazosin, which was titrated to 2 mg daily. He then underwent fine needle aspiration (FNA) of the largest left IIb lymph node. Resulting cytology revealed a less than optimal specimen with suspicion for neoplasm. Chromogranin A immunostaining of the biospecimen was increased, consistent with a diagnosis of paraganglioma. Thyroseq v3 did not identify other genetic mutations. After the above evaluation, the patient was referred to head and neck surgery and radiation oncology. The patient was presented at their tumor board, and ultimately, it was decided the mass would be difficult to resect given its location and size. The decision was made to proceed with radiation therapy. The patient was also referred to the genetics clinic with genetic testing still in process at this time.
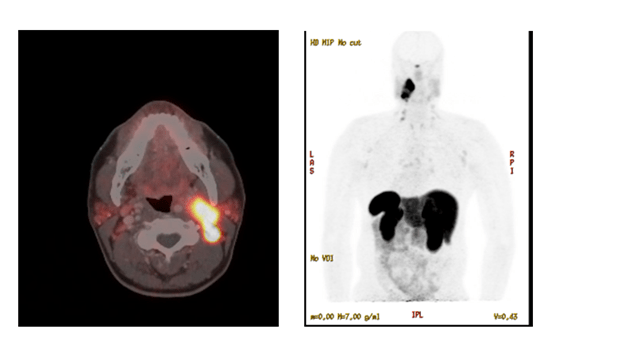
Image 2. Dotatate PET/CT scan of head and neck showing the head and neck paraganglioma.
Discussion
Paragangliomas are tumors derived from extra-adrenal chromaffin cells. They can arise from cells of the sympathetic paravertebral ganglia of the thorax, abdomen, and pelvis or parasympathetic ganglia located along the glossopharyngeal and vagal nerves in the neck and at the base of the skull. Typically, paragangliomas that arise from the parasympathetic ganglia of the head and neck are not functional, and do not produce catecholamines. Paragangliomas and pheochromocytomas (PPGLs) are often studied in conjunction, and therefore the true incidence of paragangliomas alone is unclear. The incidence of PPGLs has been estimated to be 0.2-0.8 per 100,000 people a year. Unlike for most tumors, no molecular, cellular, or histological markers are available for determining whether a PPGL is malignant. Malignancy is defined as presence of chromaffin cells in non-chromaffin organs. Most common sites of malignancy are lymph nodes, the skeleton, liver, and lungs. Depending on the study reviewed, it is estimated for PPGLs that malignancy risk is 10-17%. The biggest risk factor predisposing to malignancy is specific genetic mutations. It is estimated in 33% of PPGLs there is a genetic mutation present. For paragangliomas involving the head and neck, the following genes, SDHD, SDHB, and SDHC, VHL, and NF1 are most found to have mutations, each carrying varying degrees of malignancy risk, with SDHB carrying the highest risk, which is up to 40-80%. Classification of paragangliomas has been revised various times, with the most recent classification dividing paragangliomas into three clusters: cluster 1A/1B, cluster 2, and cluster 3. Each cluster has a different molecular pathogenesis pathway, biochemical and imaging profile, and clinical evaluation and management. For example, cluster 1A is most often associated with mutations involving the SDH genes, often linked with head and neck tumors, more likely to have a higher malignancy potential.
Clinical presentation of head and neck paragangliomas mainly result from mass effect from the location they are in, rather than from excess catecholamine secretion. If catecholamine excess is present, patients most commonly present with headaches, diaphoresis, palpitations, and hypertension. Diagnostic evaluation for PPGLs should include biochemical testing with measurement of plasma-free metanephrines or urinary fractionated metanephrines. Plasma-free metanephrines and urinary fractionated metanephrines are superior to other initial screening laboratory testing for catecholamine excess. Blood draw should occur with patients in the supine position after 30 minutes. Liquid chromatography with electrochemical or fluorometric detection (LC-ECD) and liquid chromatography with tandem mass spectrometry (LC-MS/MS) is preferred over immunoassay methods of measurement. The initial choice of imaging and location of imaging should include CT or MRI depending on which cluster of PPGL is suspected. Functional imaging such as somatostatin analogues and dotatate scans are becoming more commonly used, and have been found to have very high sensitivity and specificity for localization. The initial choice of treatment depends on size, location, biochemical status, and symptomatic nature of the tumor. For tumors with regional metastasis, such as in our case, surgery is preferred if feasible. If not feasible, radiation or ablative therapies would be an option. More targeted systemic therapies involving somatostatin radionuclide analogues and cytotoxic targeted molecular chemotherapies are also currently being studied and used more often.
Conclusion
This rare neuroendocrine tumor case highlights the initial biochemical and imaging work up done for paraganglioma, which can be complex with various confounders. It is important to keep in mind the updated classification of paragangliomas, and how each cluster corresponds to different genetic mutations. These genetic mutations will likely play a larger role in targeted treatments in the years to come.
References
1. Jacques W. M. Lenders, Quan-Yang Duh, Graeme Eisenhofer, Anne-Paule Gimenez-Roqueplo, Stefan K. G. Grebe, Mohammad Hassan Murad, Mitsuhide Naruse, Karel Pacak, William F. Young, Jr, Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline, The Journal of Clinical Endocrinology & Metabolism, Volume 99, Issue 6, 1 June 2014, Pages 1915–1942.
2. Gustavo F C Fagundes, Madson Q Almeida, Perioperative Management of Pheochromocytomas and Sympathetic Paragangliomas, Journal of the Endocrine Society, Volume 6, Issue 2, February 2022, bvac004.
3. Svenja Nölting, Nicole Bechmann, David Taieb, Felix Beuschlein, Martin Fassnacht, Matthias Kroiss, Graeme Eisenhofer, Ashley Grossman, Karel Pacak, Personalized Management of Pheochromocytoma and Paraganglioma, Endocrine Reviews, 2021;, bnab019.
4. Dana Erickson, Yogish C. Kudva, Michael J. Ebersold, Geoffrey B. Thompson, Clive S. Grant, Jon A. van Heerden, William F. Young, Jr., Benign Paragangliomas: Clinical Presentation and Treatment Outcomes in 236 Patients, The Journal of Clinical Endocrinology & Metabolism, Volume 86, Issue 11, 1 November 2001, Pages 5210–5216.
5. Plouin, P. F., Amar, L., Dekkers, O. M., Fassnacht, M., Gimenez-Roqueplo, A. P., Lenders, J. W. M., Lussey-Lepoutre, C., Steichen, O., & on behalf of the Guideline Working Group. (2016). European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma, European Journal of Endocrinology, 174(5), G1-G10.
