


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
23 Minutes
AMIT SINHA, MD
Assistant Professor, Department of Physical Medicine and Rehabilitation
University of Pittsburgh School of Medicine
LAUREN KREMM, DO
Fellow, Department of Physical Medicine and Rehabilitation
University of Pittsburgh School of Medicine
EMILY ROBBINS, DO
Resident, Department of Physical Medicine and Rehabilitation
University of Pittsburgh School of Medicine
A 5-year-old boy with a history of global developmental delay was diagnosed with influenza A per his PCP and presented to a local hospital ED with five days of lethargy, poor PO intake, decreased urine output, and fever. He was transferred to the closest specialty pediatric hospital where an MRI revealed decreased signal in the basal ganglia, and an EEG showed slowing consistent with metabolic versus infectious encephalopathy. A genetic work-up noted significant heteroplasmy for mutation of the m.3243A>G MT-ATP6 gene, which is associated with MELAS (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes) syndrome. Although a modified barium swallow study did not demonstrate any swallowing dysfunction, he fatigued quickly during mastication and was unable to meet his caloric needs by mouth. He was cleared for a pureed diet with thin liquids, but a nasogastric tube was placed to ensure that he received adequate calories and hydration. During the acute hospitalization, his exam showed dystonia, and he remained belowhis functional baseline prompting admission to the pediatric inpatient rehabilitation unit. At the time of admission, he required maximum assistance for bed mobility, transfers, and activities of daily living (ADLs). He also was not verbalizing consistently. His parents asked the UPMC pediatric rehabilitation medicine team about realistic goals and expectations for recovery.
Due to mutations in mitochondrial DNA (mtDNA) or mitochondria-related nuclear DNA (nDNA), mitochondria in affected individuals cannot appropriately generate cellular energy in the form of adenosine triphosphate (ATP). This is particularly troublesome in organs that have high-energy demands, such as the nervous system, skeletal and cardiac muscles,kidneys, liver, and endocrine glands.1 There are several identified mitochondrial disorders that may be encountered by a practicing physiatrist. In this publication, we will briefly discuss NARP, Leigh Syndrome, Alpers-Huttenlocher syndrome, and MERRF. The focus of this publication will be MELAS syndrome.
Neuropathy, ataxia, and retinitis pigmentosa (NARP) is a condition that primarily affects both the central and peripheral nervous systems with symptoms often presenting in early childhood. Affected individuals will often have weakness, numbness, and tingling and/or pain in their peripheral extremities due to sensory or sensorimotor axonal polyneuropathy. In addition, affected individuals also may have ataxia, dystonia, vision loss, learning disabilities, seizures, and cardiac arrhythmias. As affected individuals age, they may experience early onset dementia. Although onset of symptoms typically occurs early in life, individuals with NARP may not have any clinical progression of their symptoms for many years. However, affected individuals can experience episodic worsening of their symptoms or development of new symptoms, especially during times of viral illness.
NARP primarily results from mutations in the MT-ATP6 gene, which is within the mtDNA. Typically, the MT ATP6 protein is part of the enzyme ATP synthase, which is involved in the last step of mitochondrial ATP production. Mutations in the MT-ATP6 gene affect the structure or function of ATP synthase, which decreases the ability of mitochondria to make ATP. However, it remains unclear how disruption of mitochondrial ATP production exactly contributes to the progressive clinical changes described above.
Unfortunately, strict clinical diagnostic criteria for NARP has not yet been established nor is there any data on the prevalence of NARP. Molecular genetic testing for NARP can include targeted single-gene testing, mitochondrial genome sequencing, and more comprehensive whole exome testing. A discussion of specific genetic diagnostic testing is beyond the scope of this publication.
Similar to most mitochondrial disorders, there is no curative treatment for NARP. Supportive management of NARP includes management of pain, seizures, dystonia, and cardiomyopathy, as well as maximizing function through therapy and assistive devices. It should be noted that sodium valproate and barbiturates should be avoided for seizure management in this population, as these medications have an inhibitory effect on the mitochondrial respiratory chain. There continues to be ongoing research regarding the potential use of antioxidants, gene therapy, vitamins, and other compounds as treatment modalities for NARP.2
Leigh Syndrome, often referred to as subacute necrotizing encephalomyelopathy, is a progressive neurometabolic disorder often characterized by the degeneration of the central nervous system. This degenerative disease can occur because of mutations to either nDNA or mtDNA. The mutations can result in deficiencies of enzymes that affect the mitochondrial respiratory chain complexes or the pyruvate dehydrogenase complexes, resulting in decreased energy production.
The onset of symptoms can occur in early infancy, often within the first year of life, and progresses rapidly. The first signs of Leigh Syndrome are often neurodevelopmental regression, poor sucking, dysphagia, and disruption in eating patterns resulting in a failure to thrive. In addition to the early clinical signs, Leigh Syndrome should be suspected when symptoms associated with basal ganglia, cerebellar, or brainstem disease are present (i.e., ataxia, ophthalmoparesis, nystagmus, optic atrophy, respiratory failure, dystonia, and hypotonia). Other clinical features include seizures and peripheral neuropathy.
In addition to clinical features, elevated lactate levels in the blood and cerebrospinal fluid (CSF) are often present. Hyperlactatemia can further be confirmed by elevated levels of alanine concentrations in plasma amino acids. Radiographic findings on brain imaging can be seen on both CT and MRI. CT findings may demonstrate bilateral hypodensities in the basal ganglia, while MRI may demonstrate bilateral hyperdensities of the brainstem or basal ganglia.
There is no known cure for Leigh Syndrome and death typically occurs within two to three years because of respiratory failure. Treatment includes symptom management and surveillance.2,3
Alpers-Huttenlocher syndrome (AHS) is a mitochondrial disorder that is one of the most severe of a group of conditions known as POLG-related disorders. AHS is caused primarily by mutations in the POLG gene, which is responsible for mtDNA replication and repair. Individuals with AHS typically have three characteristic clinical findings: intractable epilepsy, psychomotor regression, and liver disease. Onset of these clinical findings most commonly occurs between the ages of 2 and 4 years, although there is a second peak of onset between the ages of 17 and 24 years. Overall prevalence is estimated to be 1:100,000 live births. Children with AHS are typically healthy and develop appropriately until disease onset. Seizures constitute the initial presentation in about 50 percent of affected individuals. After onset of seizures, the disease progresses quickly, with death usually occurring within four years of onset. However, there are several cases where disease progression is slower. In addition to seizures, patients may also demonstrate progressive ataxia, visual loss, cranial nerve dysfunction, respiratory insufficiency, movement disorders, and sensorimotor axonal polyneuropathy, which can lead to hypotonia and areflexia. Like other mitochondrial disorders, viral infections often trigger onset of symptoms and exacerbate symptom progression.
At the first onset of symptoms, 70 percent of affected individuals have a normal range of mtDNA copy number in their liver and muscle tissue. However, as the disease progresses, as mtDNA is unable to successfully replicate, mtDNA copy number decreases, eventually leaving affected individuals with < 35 percent of normal levels of mtDNA in their liver and muscle tissue, likely contributing to a reduction in cellular energy. For this reason, AHS is classified as an mtDNA-depletion syndrome.4
Myoclonic Epilepsy with Ragged Red Fibers (MERFF) is a multi-system disorder that often presents in childhood or adolescence from mutations to nDNA or mtDNA. The most common genetic mutation occurs at the MT-TK gene, while less commonly, mutations to the MT-TL1, MT-TH, and MT-TS1 genes are seen. Mutations of the MT-TL1, MT-TH and MT-TS1 genes result in a mixed clinical picture of MERFF plus additional symptoms of other mitochondrial disorders.
Symptom onset typically occurs after a period of normal early development. The most common initial symptom is myoclonus followed by generalized epilepsy, ataxia, weakness, and dementia.
Additional clinical findings associated with MERFF may include: sensorineural hearing loss, myopathy, peripheral neuropathy, short stature, optic atrophy, cardiomyopathy with Wolff-Parkinson-White Syndrome (WPW) Syndrome, pigmentary retinopathy, and lipomas.
The diagnosis of MERFF requires the presence of myoclonus, epilepsy, and ragged red fibers on muscle biopsy or the presence of a MERFF clinically significant nDNA or mtDNA mutation on genetic testing. Additional testing to further support the diagnosis of MERFF can include elevated serum or CSF lactate and elevated CSF protein. Cardiac and neurological testing can further identify sequelae of the disease.
There is no known cure for MERRF. Standard treatment includes prescribing Coenzyme Q10 and L-carnitine to preserve mitochondrial function. Additional treatment includes symptom management and surveillance.5
MELAS syndrome, which was first delineated in 1984, is the most common maternally inherited mitochondrial disorder.6 Diagnostic criteria for MELAS syndrome were published in 1992 with the following three criteria: 1) stroke-like episodes before 40 years of age, 2) encephalopathy characterized by seizures and/or dementia, and 3) mitochondrial myopathy showing lactic acidosis and/or ragged red fibers. Furthermore, the diagnosis may be confirmed if at least two of the following three criteria are met: 1) normal early psychomotor development, 2) recurrent headaches, and 3) recurrent episodes of emesis.7 The prevalence of MELAS syndrome remains unclear. An epidemiologic study reviewing occipital strokes in individuals between the ages of 18 to 45 years completed in northern Finland estimated that the prevalence of the m.3243A>G mutation was > 16.3:100,000. This study suggested that six percent of patients less than 45 years of age and 14 percent of patients less than 30 years of age had the m.3243A>G mutation.8 Another more recent epidemiologic study completed in Japan estimated the prevalence of MELAS to be 0.2:100,000.9 Although this remains an uncommon diagnosis, pediatric physiatrists and other specialists in tertiary or quaternary specialty pediatric hospitals will most likely encounter children with MELAS syndrome.
Mitochondrial dysfunction is due to inherited or point mutations involving either mtDNA or, more commonly in children, nDNA. mtDNA is a circular DNA comprised of 16,596 base pairs with 13 genes encoding subunits of the respiratory chain, 22 transfer- RNA, and two ribosomal RNA.10,11 The presence of normal and mutated mtDNA in various frequencies in different cell populations is known as heteroplasmy. Exceeding a critical level for a specific mutation allows for symptom manifestation, although this critical level may vary in different tissues and between different mitochondrial disorders. The mtDNA genetic alterations are inherited in a maternal fashion and directly affect the mitochondrial 5-complex respiratory chain. The respiratory chain (RC) is the end pathway of aerobic metabolism and is directly involved inoxidative phosphorylation needed to create energy in the form of adenosine triphosphate (ATP).12 These mutations can result in impaired synthesis of RC subunits, causing energy deficiency and organ failure.
MELAS is specifically associated with at least 39 mtDNA mutations. The most common inherited mutation in MELAS syndrome is the leucine tRNA point mutation m.3243A>G located on the MT-TL1 gene of mtDNA.1,10,13 The pathophysiology of MELAS is not fully defined. There are multiple mechanisms that are thought to play a role in the multi-organ dysfunction. The first theory couples impaired energy production with microvascular angiopathy. Impaired synthesis of RC complexes results in insufficient mitochondrial energy production, which in turn contributes significantly to the multi-organ dysfunction observed in MELAS syndrome.10,13 This energy deficiency triggers a reflexive mitochondrial proliferation consisting of predominantly dysfunctional, enlarged mitochondria with complicated cristae that are unable to overcome the impaired synthesis of RC complexes. This proliferation induces microvascular angiopathy when occurring in endothelial cells of small blood vessels. It is speculated that microvascular angiopathy plays a critical role in the stroke-like episodes of MELAS syndrome.1,10,14,15
A second theory proposes that a deficiency of nitric oxide (NO) is present in patients with MELAS syndrome. Citrulline, a precursor to NO, is produced in the mitochondria. Citrulline concentrations are reduced in the setting of mitochondrial dysfunction, resulting in decreased NO. A normal volume of NO allows vascular smooth muscle relaxation, promoting adequate vascular perfusion. Decreased NO is theorized to result in decreased perfusion of multi-organ systems in MELAS, resulting in organ dysfunction.1,13,14,16,17
Mitochondrial dysfunction is represented as a complex group of disorders that can impact multiple organ systems. Groups of symptoms involving multiple organ systems are often indicative of distinct clinical syndromes. However, it is not uncommon for patients to present with an unclear pattern of symptom pathology.18
Myopathy is the most common symptom associated with mitochondrial disorders. Neck flexors are affected early, followed by neck extensors with progression to proximal muscle groups.7 Other generalized symptoms include: premature fatigue, exercise intolerance, myalgias, ophthalmoplegia, hearing loss, diabetes mellitus, and thyroid disorders.1,16,18
MELAS syndrome is a multi-organ disease. Greater than 50 percent of patients present before the age of 10, and > 70 percent by the age of 20.10,14 Evolution of symptoms occurs after a period of normal or expected development.
The hallmark manifestations associated with MELAS syndrome are myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. Additional symptoms to confirm the diagnosis include normal early psychomotor development, recurrent headaches, or recurrent vomiting.1,16
The summarization of manifestation frequency at disease onset and throughout the clinical course of MELAS syndrome are noted in Table 1.1
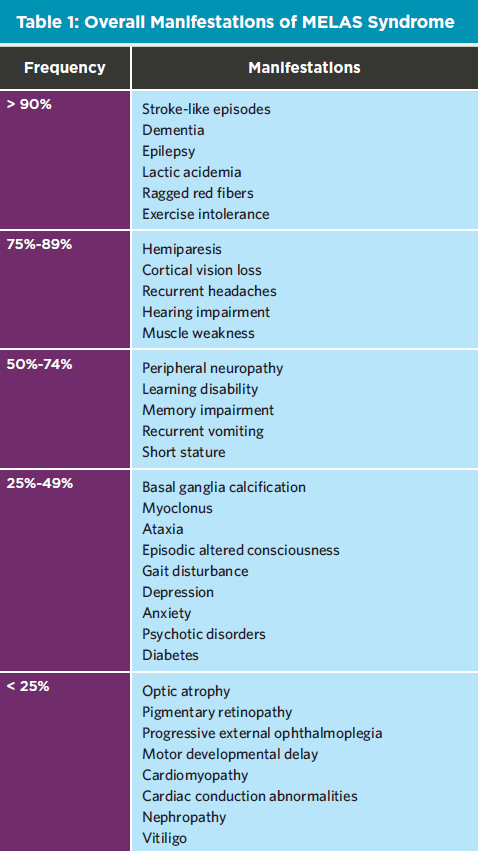
Recurrent headaches occur early, but later in the disease headaches can become a precursor to seizures and/or stroke-like episodes. Seizures can occur independently or be correlated to stroke-like episodes.1,14 Stroke-like episodes with cortical visual changes, aphasia, and transient hemiparesis are a cardinal symptom required for clinical diagnosis.1,18 Each stroke-like episode is usually followed by a remission; however, each new episode has a cumulative negative effect. Over time, stroke-like episodes can contribute to progressive dementia.1,16,18
Additional multi-system clinical manifestations may include cardiomyopathy (50 percent of cases), cardiac conduction abnormalities, diabetes mellitus type I or II, gastrointestinal alterations, peripheral neuropathy, movement disorders, hearing impairment, myoclonus, ataxia, optic atrophy, psychiatric abnormalities, and ophthalmoplegia.1,19 Lactic acidosis is present in 94 percent of individuals with MELAS syndrome and is a key diagnostic component.1,18 Differential Diagnosis It is important to recognize the multi-organ system presentation to distinguish MELAS syndrome from other diagnoses. Common mimics include acute stroke, myasthenia gravis, myotonic dystrophy, multiple sclerosis, metabolic disease, and migraines.
Motor Weakness and Exercise Tolerance
Locomotor training with body weight support is an effective way to improve ambulation, however these results have been demonstrated only in case studies and with small sample sizes.20 Due to exercise intolerance, many individuals with MELAS syndrome adopt a sedentary lifestyle, which leads to further deconditioning. Deconditioning has been associated with decreases in mitochondrial numbers and respiratory chain activity in skeletal muscle, as well as reduced cardiovascular activity. It has been postulated that deconditioning in individuals with mitochondrial disorders may lead to a decrease in functional mitochondria levels.21
Studies have shown that endurance training in individuals with mitochondrial myopathy is well tolerated, increases exercise capacity, and improves the quality of life.22-24 However, the relative proportion of mutant mtDNA appears to increase in individuals with mitochondrial myopathies who participated in endurance training. This has raised concerns about the safety of endurance training, because the clinical progression of mtDNA diseases is related to the progressive accumulation of mutant mtDNA in various tissues. Yet, in this same population, endurance training increased absolute levels of wild-type mtDNA, which may explain why endurance training generated improvement in exercise capacity.21 Further research is needed to definitively show the safety and efficacy of endurance training in MELAS syndrome.
Evidence to support resistance training is also inconclusive. In a single-case report, resistance training yielded a decrease in the proportion of mutant relative to wild-type mtDNA.25 A subsequent pilot study with a small group of subjects performing unilateral leg resistance training demonstrated an improvement in muscle strength in all subjects and increased muscle enzymatic activity in two of three subjects.26
Ataxia
Synofzik and Ilg found that traditional physical therapy for ataxia may be improved by therapies involving video game technology. The video games encouraged multi-joint coordination, dynamic stability, and increased rapid movements needed to compensate for gait perturbations that contribute to falls.27 Locomotor training with body weight support and over-ground gait training also are routinely used for the treatment of ataxia. Unfortunately, there is insufficient literature describing long-term outcomes regarding ataxia in individuals with MELAS syndrome.
Movement Disorders
Myoclonus and dystonia have been reported in individuals with MELAS syndrome.Other movement disorders, such as parkinsonism, tremor, and chorea are rarer but have been reported.28 In our experience, myoclonus, dystonia, and spasticity can be treated similarly to other patient populations we encounter in our clinical practice. Baclofen is our initial drug of choice, with the recommendation of starting at a conservative dose (0.5-1 mg/kg/day in children given in two to three divided doses) and titrating slowly every five to seven days based on effect while closely monitoring for adverse effects. It is imperative to keep in mind that individuals with MELAS syndrome have underlying myopathy that may become more apparent with upward titration of baclofen. Clonazepam is our initial drug of choice for myoclonus, starting with a very conservative dose (0.01-0.03 mg/kg/day given in two to three divided doses) and titrating slowly. We would recommend discussion with the patient’s primary genetic and/or metabolic physician prior to beginning any of these medications. Individuals with MELAS syndrome are at risk of developing contractures, particularly if their mobility is limited and/or they have developed spasticity or dystonia. Tendon stretch may be achieved using stretching orthotics that are used throughout the day and/or night if they do not interfere with sleep.
Dysphagia
de Laat et al. suggested that the incidence of dysphagia can range from 18 percent to 45 percent.29 The weakness of the lips, tongue, or jaw results in impairment of bolus preparation. As such, this increases bolus transit time, which may cause pharyngeal pooling that increases risk for aspiration.26 In these patients, solid foods cause more problems than liquids. Interestingly, the typical prescription for dysphagia patients includes thickened liquids, which may worsen function in this population as these liquids take more energy to consume. Because dysphagia typically develops slowly and is not severe, patients can develop adaptations for safe and efficient swallowing. Therapeutic exercises that focus on improving jaw closure, increasing tone in the posterior sections of the tongue, and improving rotary chewing can help swallowing function.30 For patients with insufficient oral intake secondary to dysphagia or fatigue, a nasogastric or percutaneous endoscopic gastrostomy tube may be required for nutritional supplementation.
Nutritional Considerations
Recent studies have suggested that supplementation with citrulline and arginine, the NO precursors, can improve the availability of NO in the blood stream, which in turn improves perfusion. The use of arginine has been more widely studied and has been associated with a decrease in the severity and frequency of stroke-like episodes in MELAS syndrome.1,14 The recommended dosing of L-arginine during acute stroke-like episodes in children is an intravenous bolus dose of 500 mg/kg followed by a continuous 24-hour infusion of 500 mg/kg for approximately three to five days. The recommended prophylactic maintenance dose for children after the initial stroke-like episode is 150 to 300 mg/kg divided into three doses.1,17
Creatine and coenzyme Q10 (CoQ10) have also been used in MELAS syndrome. The by-products of creatine act as a phosphate donor for ATP production, while CoQ10 facilitates electron transfer and stabilization of respiratory chain complexes within the mitochondria. The recommended dosing of creatine and CoQ10 for children are 100 mg/kg/day and 5 to 10 mg/kg/day, respectively.
The progression of MELAS syndrome and other mitochondrial disorders is variable; however, one commonality is that poor survival has been seen in patients with onset before 6 months of age and a higher plasma lactate level.31 As the disorder progresses, patients may develop neurologic deterioration such as progressive dementia secondary to the recurrent stroke-like episodes and seizures. Of note, Testai found that a younger age of onset has been shown as an independent predictor of death.32 In a natural history study following 31 individuals with MELAS syndrome and 54 symptomatic and asymptomatic carriers over 10.6 years, neurologic examination, cognitive testing, and daily living measures significantly declined in all those with MELAS syndrome but not in carriers. In this study, the average age of death in those with MELAS syndrome was 34.5 + 19 years (range 10.2-81.8 years), with 22 percent of deaths occurring in those under 18 years of age.33 In another natural history study done in Japan following 96 individuals with MELAS syndrome, 20.8 percent of affected individuals died within a median time of 7.3 years from diagnosis.9
Rehabilitation management, including nutritional supplementation, anti-spasticity medications, enteral supplementation, orthoses and assistive devices, as well as physical, occupational, and speech therapy, all serve to maximize the function of the patient. These interventions minimize further illness and disability, and in some cases prevent recurrence of stroke-like episodes. There is no evidence that any of the interventions affect mortality of MELAS syndrome, but they reduce the morbidity associated with the progressive nature of this disease.
Functional prognosis of pediatric patients with mitochondrial disorders ranges from independent to total dependence. Most children require maximal assistance to total dependence for mobility, and assistive devices, including walkers, orthoses and wheelchairs, are required for most children. Many patients are dependent on their parents or other caregivers for the duration of their life. The Pediatric Evaluation of Disability Inventory questionnaire, used to assess the extent of functional impairment, identified complex functional limitations in everyday life involving motor and sensory function, communication, and intelligence.34
Despite making gains in taking nutrition by mouth, a g-tube was placed for insufficient caloric and hydration intake. The patient made functional gains in his ability to participate in ADLs, including supervision for dressing and minimal assistance for self-feeding. He also made improvements in fine motor skills, such as being able to stack blocks and color. He could take steps with close supervision, but he continued to require a gait belt for safety due to his ataxia. The patient’s parents could fully understand his speech, while noncaregivers could understand ~75 percent of spoken words. Throughout his stay, he was followed closely by the inpatient genetics consult team (including a genetic nutritionist) who monitored laboratory results, including serial serum lactic acid levels, and assisted with nutrition supplementation. He was ultimately discharged home with his family with outpatient physical, occupational, and speech therapy and a plan to start half-day kindergarten. References
1. El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS Syndrome: Clinical Manifestations, Pathogenesis and Treatment Options. Mol Genet Metab. 2015; 116: 4-12.
2. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. GeneReviews® [Internet], U.S. National Library of Medicine, 28 Sept 2017; https://www.ncbi.nlm.nih.gov/pubmed/20301352.
3. Leigh Syndrome. Genetics Home Reference. June, 2016; https://ghr.nlm.nih. gov/condition/leigh-syndrome.
4. Saneto RP, Cohen BH, Copeland WC, Naviaux RK. Alpers-Huttenlocher Syndrome. Pediatr Neurol. 2013; 48:1 67-78.
5. DiMauro S, Hirano M. MERRF. GeneReviews® [Internet]. U.S. National Library of Medicine, 29 Jan 2015; www.ncbi.nlm.gov/books/NBK1520/.
6. Pavlakis SG, Phillips PC, DiMauro S, DeVivo DC, Rowland LP. Mitochondrial Encephalopathy, Lactic Acidosis and Stroke-like Episodes: A Distinctive Clinical Syndrome. Ann Neurol. 1984; 16(4): 481-8.
7. Hirano M, Ricci E, Keonigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, DiMauro S, Rowland LP. MELAS: An Original Case and Clinical Criteria for Diagnosis. Neuromuscul Disord. 1992; 2(2): 123-35.
8. Majamaa K, Turkka J, Karppa M, Wingvist S, Hassinen IE. The Common MELAS Mutation A3243G in Mitochondrial DNA Among Young Patients With an Occipital Brain Infarct. Neurology. 1997; 49(5): 1331-4.
9. Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, Kakuma T, Koga Y, Taro Matsuoka for MELAS Study Group in Japan. MELAS: A Nationwide Prospective Cohort Study of 96 Patients in Japan. Biochim Biophys Acta. 2012; 1820(5): 619-24.
10. Debray FG, et al. Disorders of Mitochondrial Function. Curr Op Pediatr. 2008; 20(4): 471-82.
11. Neupert W, Herrmann JM. Translocation of Proteins Into Mitochondria. Ann Rev Biochem. 2007; 76: 723-49.
12. Falk MJ. Neurodevelopmental Manifestations of Mitochondrial Disease. J Dev Behavior Pediatr. 2010; 31(7): 610-21.
13. Koga Y, et al. Molecular Pathology of MELAS and l-Arginine Effects. Biochim Biophy Acta (BBA) - General Subjects. 2012; 1820(5): 608-14.
14. El-Hattab AW, et al. Impaired Nitric Oxide Production in Children With MELAS Syndrome and the Effect of Arginine and Citrulline Supplementation. Mol Genet Metab. 2016; 117(4): 407-12.
15. Sproule DM, Kaufmann P. Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes: Basic Concepts, Clinical Phenotype, and Therapeutic Management of MELAS Syndrome. Ann N Y Acad Sci. 2008; 1142: 133-58.
16. DiMauro S. “MELAS”. GeneReviews® [Internet]. U.S. National Library of Medicine, 21 Nov. 2013; www.ncbi.nlm.gov/books/NBK1233/.
17. El-Hattab AW, Almannai M, Scaglia F. Arginine and Citrulline for the Treatment of MELAS Syndrome. J Inborn Errors Metab Screen. 2017; 5.
18. Alexander MA, et al. Pediatric Rehabilitation: Principles and Practice. Demos Medical. 2015.
19. Lorenzoni PJ, Werneck LC, Kay CS, Silvado CE, Scola RH. When Should MELAS (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes) Be the Diagnosis? Arq Neuropsiquiatr. 2015; 73(11): 959-67.
20. Cernak K, Sevens V, Price R, Shumway-Cook A. Locomotor Training Using Body-Weight Support on a Treadmill in Conjunction With Ongoing Physical Therapy in a Child With Severe Cerebellar Ataxia. Physical Therapy. 2008; 88(1): 88-97.
21. Taivassalo T, Haller RG. Implications of Exercise Training in mtDNA Defects – Use It or Lose It? Biochim Biophys Acta. 2004; 1659(2-3): 221-31.
22. Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DA, Haller RG. Aerobic Conditioning in Patients With Mitochondrial Myopathies: Physiological, Biochemical and Genetic Effects. Ann Neurol. 2001; 50: 133-41.
23. Taivassalo T, Matthews PM, DeStefano N, Sripathi N, Genge A, Karpati G, Arnold DL. Combined Aerobic Training and Dichloroacetate Improve Exercise Capacity and Indices of Aerobic Metabolism in Muscle Cytochrome Oxidase Deficiency. Neurology. 1996; 47: 529-34.
24. Taivassalo T, DeStefano N, Argov Z, Matthews PM, Chen J, Genge A, Karpati G, Arnold DL. Effects of Aerobic Training in Patients With Mitochondrial Myopathies. Neurology. 1998; 50: 1055-60.
25. Taivassalo T, Fu K, Johns T, Arnold D, Karpati G , Shoubridge EA. Gene Shifting: A Novel Therapy for Mitochondrial Myopathy. Hum Mol Genet. 1999; 8: 1047-52.
26. Taivassalo T, Shoubridge E, Wyrick P, Lofberg M, Haller RG. Resistance Exercise Training for Mitochondrial Myopathies: Effects on Muscle Strength and Mitochondrial Function. Med Sci Sports Exerc. 202; 34: A943 (Abstract).
27. Synofzik M, Ilg W. Motor Training in Degenerative Spinocerebellar Disease: Ataxia-Specific Improvements by Intensive Physiotherapy and Exergames. Biomed Res Int. 2014; April 27.
28. Tranchant C, Anheim M. Movement Disorders in Mitochondrial Diseases. Rev Neurol (Paris). 2016; 172(8-9): 524-9.
29. de Laat P, Zweers HEE, Knuijt S, Smeitink JAM, Wanten GJA, Janssen MCH. Dysphagia, Malnutrition and Gastrointestinal Problems in Patients With Mitochondrial Disease Caused by the M3243a>G Mutation. Neth J Med. 2015; 73(1): 30-36.
30. Vandana VP, Bindu PS, Sonam K, Taly AB, Gayathri N, Madhu N, Sinha S. Speech-language and Swallowing Manifestations and Rehabilitation in an 11-year-old Girl With MELAS Syndrome. J Pediatr Neurosci. 2015; Jan-Mar 10(1):31-4.
31. Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir LD, Tzoulis C, Uusimaa J, De Angst IB, Lönnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, Darin N. A Multicenter Study on Leigh Syndrome: Disease Course and Predictors of Survival. Orphan J Rare Dis. 2014; 9: 52.
32. Testai FD, Gorelick PB. Inherited Metabolic Disorders and Stroke Part 1; Fabry Disease and Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Strokelike Episodes. Arch Neurol. 2010; 67(1): 19-24
33. Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM, et al. Natural History of MELAS Associated With Mitochondrial DNA m.3243A>G Genotype. Neurology. 2011; 77(22): 1965-71.
34. Rognac M, Meznaric M, Zeviani M, Sperl W, Neubauer D. Functional Outcome of Children With Mitochondrial Diseases. Pediatr Neurol. 2011; 44(5): 340-346.
