


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
5 Minutes
This case was conducted by Year II Gastroenterology Fellow, Nelson Valentin, MD, MSc.
An 18-year-old female with no significant past medical history presented with one week of general malaise, nausea, vomiting, and jaundice. Aside from intermittent migraines, for which she takes sumatriptan as needed, she was in her usual state of health up until presentation. She took no other medications regularly. She endorsed occasional alcohol use but denied tobacco use or illicit drugs. She had recently self-quarantined for possible COVID-19 exposure but never developed symptoms.
At her initial presentation, her blood work and liver function tests were notable for a total bilirubin of 15.7 mg/dL and direct bilirubin of 8.9 mg/dL, ALT 9 IU/L, AST 115 IU/L, ALP 18 IU/L. Within 48 hours of presentation, her blood work worsened with a total bilirubin of 44.4 mg/dL, ALT <5 IU/L, AST 95 IU/L, ALP 10 IU/L. In addition, her evaluation was notable for Coombs-negative hemolytic anemia, low ceruloplasmin, and coagulopathy with an INR of 2.5. Her mental status remained intact, but she exhibited asterixis. A comprehensive viral serology workup was negative. She underwent a right upper quadrant ultrasound, with Dopplers showing a normal liver with patent hepatic vessels. A contrasted CT abdomen and pelvis showed a normal liver with mild splenomegaly. Given concerns for acute liver failure, she was transferred to a tertiary center for an expedited liver transplant evaluation.
This patient was diagnosed with Wilson’s disease (WD), a rare, genetic disorder that causes excessive copper accumulation in the liver, brain, and other organs. Normal copper intake is 2-5 mg/day, of which 40%-60% is absorbed by the stomach and duodenum via portal circulation. The copper is mainly bound to ceruloplasmin in the liver. Ceruloplasmin-bound copper is secreted into the plasma where it plays a major role in Fe2+ oxidation in plasma. After the ceruloplasmin is broken down, unbound copper binds to bile proteins and is excreted in bile.
In Wilson’s disease, there is a mutation in the ATP7B gene that encodes a copper transport ATPase. This leads to issues with incorporating the copper in ceruloplasmin, as well as the liver’s ability to excrete copper in bile. Clinical features can be highly variable and can range from asymptomatic to acute liver failure. There is a wide age spectrum of involvement between five and 55 years of age, with neurologic manifestations more common in later disease. Kayser-Fleischer (KF) eye rings are present in only 40%-60% of patients and usually present with concomitant liver disease. See Figure 1 for a diagnostic template to support Wilson’s disease treatment paradigms.
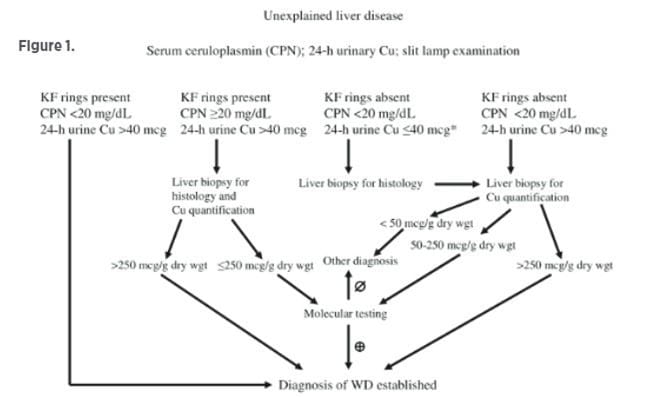
Patients will typically require chelator treatments for one to five years before going on maintenance therapy. Pharmacological therapy may include chelating therapies, such as D-penicillamine or trientine, or zinc. Neurological deterioration can occur with side effects such as fever, rash, aplastic anemia and other blood disorders, retinitis, gastritis, and possible changes in immune function. Switching to zinc monotherapy for maintenance can be beneficial, since zinc may be more specific to binding copper than chelators. Patients should avoid copper-enriched foods, such as shellfish, chocolate, mushrooms, etc. Patients typically require lifelong maintenance therapy, and treatment monitoring should be performed at least twice per year.
In patients with Wilson’s disease, progression to acute liver failure (ALF) is rare and accounts for only 2%-3% of all U.S. ALF cases. However, ALF can be the first presentation of the disease, especially in young patients. Early identification is critical, since fulminant Wilson’s disease is considered fatal without liver transplantation. As such, patients with fulminant Wilson’s disease qualify as status 1A for liver transplantation as per UNOS criteria. Please see Figure 2 for a diagnostic approach to fulminant Wilson’s disease.
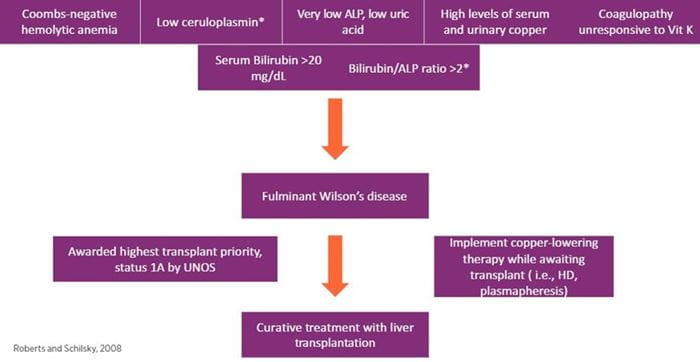
Our patient met criteria for fulminant Wilson’s disease given her presentation and laboratory findings highly suggestive of WD. She was listed as a status 1A for liver transplantation within 24 hours of presentation to UPMC. The patient received an offer within 24 hours and was transplanted four days after initial presentation. The liver explant had copper deposition consistent with WD. The patient was treated with five doses of penicillamine after transplantation, and her postoperative course was without complications. Following basiliximab induction and a routine steroid taper, the patient was started on tacrolimus and mycophenolate mofetil. The patient was discharged home on postoperative day nine.
Wilson’s disease is a rare cause of ALF in the United States, but a high clinical suspicion is warranted. Fulminant Wilson’s disease is fatal without liver transplantation. In many cases, serum copper and 24-hour urinary excretion of copper may not be readily available, so practitioners need to rely on other clinical features. Medical professionals should consider Wilson’s disease if a young patient presents with very high bilirubin and low ALP (typically <40 IU/L) in the absence of other indicators. Fulminant Wilson’s disease qualifies as a status 1A priority listing for UNOS, and liver transplantation is curative for Wilson’s disease.
Roberts EA, Schilsky ML, American Association for the Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008 Jun;47(6):2089-111. PMID: 18506894.
Lau JY, Lai CL, Wu PC, Pan HY, Lin HJ, Todd D. Wilson’s disease: 35 years’ experience. Q J Med. 1990 Jun;75(278):597-605. PMID: 2217665.
Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology. 1991 Apr;100(4):1129-34. PMID: 2001814.
