


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
9 Minutes
A 28-year-old Asian female presented during pregnancy with elevated, difficult to control blood pressure (BP), leading to an induced delivery of a preterm 32-week-old infant. The infant was admitted to the neonatal intensive care unit (NICU), where she was hospitalized for three weeks with eventual discharge to home in good health.
A few weeks following the delivery, the patient (the mother) was evaluated in the endocrine clinic. She reported a history of episodic pulsatile headaches, palpitations, and excessive sweating for the past year. Each episode lasted approximately 10 minutes and resolved spontaneously. Her systolic blood pressure (SBP) was elevated during these episodes (200 mm/Hg) and would subsequently return to normal following the episode. Antihypertensive treatment was not started during or following the pregnancy.
There was no history of prior hospitalizations or surgeries. She never used cigarettes. The patient reported that her mother had died suddenly in her 30s from an unknown cause. The patient’s physical exam was unremarkable at the time of her endocrine evaluation. Her BP was 105/76 at the time of her visit.
An evaluation for secondary causes of hypertension was initiated. Thyroid function studies, aldosterone, plasma renin activity, adrenocorticotropic hormone, and cortisol levels all were within normal limits. Plasma and urine metanephrines and normetanephrines were elevated (Table 1). Chromogranin A also was elevated.
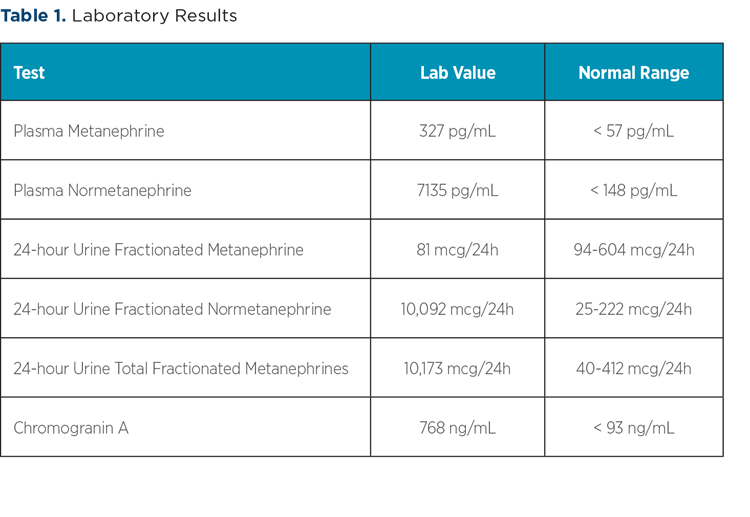
A computed tomography (CT) scan of the chest, abdomen, and pelvis demonstrated a large right retroperitoneal, heterogeneously enhancing mass, measuring 5.8 x 3.7 x 8.3 cm, which encircled the aorta approximately 180 degrees. There was no evidence of vascular invasion (Figure 1).
In preparation for surgical excision of this mass, treatment with diltiazem 60/30/60 mg three times a day was initiated for preoperative BP control. Two weeks after diltiazem initiation, the patient underwent laparoscopic resection of the tumor. Surgical pathology showed a 6.8 cm mass with lesional cells that were positive for synaptophysin and chromogranin. Staining was negative for AE1/3 and sustentacular cells that were highlighted by S100. These results were consistent with a diagnosis of paraganglioma.
Postoperatively, the patient was weaned from antihypertensive medications for two weeks. She did not experience postoperative pain. Two weeks after surgery, repeat plasma metanephrines were 26 pg/mL and normetanephrines 164 pg/mL.
The patient agreed to proceed with genetic testing in the UPMC Endocrine Genetics Clinic to determine a potential hereditary cause for the development of paragangliomas and pheochromocytomas. This panel included testing for mutations in the SDHx genes, Von Hippel-Lindau syndrome (VHL), Multiple Endocrine Neoplasia type 1 (MEN1), Multiple Endocrine Neoplasia type 2 (MEN2), and neurofibromatosis type 1 (NF1).
The genetic testing was positive for a pathogenic heterozygous mutation (deletion of Exon 1) of the SDHB gene, which is associated with autosomal dominant pheochromocytoma-paraganglioma syndrome. Due to these results, genetic testing is planned for family members, including the patient’s daughter and sister.
The finding of a paraganglioma or pheochromocytoma (PGL/PHEO) during pregnancy is a rare diagnosis. The prevalence is reported to be less than one case in 50,000 full-term pregnancies.1 In approximately 20% of the cases, the diagnosis is made during the postpartum period, increasing the risk of potentially unfavorable pregnancy outcomes. Timely diagnosis decreases the maternal mortality rate from 40% to 5% and fetal mortality rate from 50% to 15%.2
The diagnosis of PGL/PHEO during pregnancy is challenging due in part to its rare occurrence, as well as the similarity of presentation to other, more frequent pregnancy-related hypertensive disorders. Gestational hypertension and preeclampsia/eclampsia are more commonly observed causes of hypertension during pregnancy. Paroxysmal episodes of elevated BP, occurring before 20 weeks of gestation, without edema or proteinuria, can serve to alert clinicians caring for women during pregnancy to consider a diagnosis of PGL/PHEO. A personal or family history of NF1, MEN2, or VHL makes a diagnosis of PGL/PHEO in pregnancy more likely. Patients may become increasingly symptomatic as the pregnancy progresses, and 50% of patients also can experience hypotensive episodes.2
When PGL/PHEO is suspected, the diagnosis must be confirmed by the demonstration of increased plasma or urinary metanephrines and normetanephrines. These tests have a sensitivity of 98% with a high negative predictive value, allowing exclusion of the diagnosis if results are in the normal range.1
A healthy pregnancy does not significantly affect plasma catecholamine levels, which are only slightly elevated in the setting of preeclampsia.3 False-positive results can occur with improper blood sampling (e.g., failure to rest a patient for 30 minutes in quiet surroundings) and with the use of β-adrenergic receptor blockers or tricyclic antidepressants.1
Once the diagnosis of PGL/PHEO is established with appropriate hormonal studies, tumor localization is the next step in the evaluation. Magnetic resonance imaging (MRI) without contrast is the modality of choice to diagnose PGL/PHEO in pregnancy, with sensitivity similar to that of CT scanning (90% to 100%).4 CT and MIBG scans are contraindicated during pregnancy.1-3 The patient in this case was diagnosed following delivery, which allowed for CT scanning to be performed. Although abdominal ultrasound is safe and affordable, its diagnostic sensitivity for small tumors is limited; thus, a normal test result does not always exclude the diagnosis.4
Once imaging studies confirm the presence of a mass suggestive of PGL/PHEO, surgery will be required as part of the treatment regime. The timing of surgery depends on many factors, including gestational age (if the patient is pregnant), location of the tumor, and adequate preoperative α-blockade. In women diagnosed during pregnancy, if an adequate α-blockade can be established before 24 weeks of gestation, laparoscopic surgical resection is recommended in the second trimester.3 Otherwise, medical treatment is preferred, and laparoscopic resection is postponed until after delivery of the child.
Management of elevated HTN in pregnant patients with PGL/PHEO is challenging. Although the placenta shields the fetus from high circulating levels of catecholamines, the utero-placental circulation can still be compromised due to severe arterial vaso-constriction.4 Treatment with α-adrenergic receptor blockers can counteract these effects but also can increase the risk of hypotension, which can further compromise utero-placental circulation. Phenoxybenzamine and doxazosin are the usual first therapeutic options for patients presenting with elevated BP during pregnancy who are suspected of having PGL/PHEO.5 Both medications can cross the placenta and are considered fetal risk category C, which categorizes these medications as generally safe to use during pregnancy with careful monitoring. Neonatal hypotension and respiratory depression have been reported with phenoxybenzamine, but not with doxazosin.5 Therefore, close neonatal monitoring during the first days of life is advised if a mother has been treated with phenoxybenzamine.
Catecholamine induced tachycardia and α-blockade can induce a reflex tachycardia that can be treated with β-blockers if this becomes necessary.1 Beta blockers have been associated with intrauterine growth restriction. Therefore, short-term use with the lowest possible dose is advised.2 Initiation of a β-blocker should occur only after an appropriate α-adrenergic blockade is achieved. This time lag is necessary to prevent precipitation of a hypertensive crisis.1 If β-blockers are contraindicated or fail to control blood pressure, and there is an absence of tachycardia, calcium channel blockers can be added to the treatment regimen. These medications also are categorized as fetal risk category C.
For pregnant patients with PGL/PHEO, there is no clear consensus regarding the preferred route for delivery. The elective Caesarean section was long favored based on prior case reports that suggested higher maternal mortality rates with vaginal delivery.3 More recent studies6 describe vaginal delivery as both acceptable and safe provided that there is adequate epidural analgesia to minimize pain and stress and by reducing the second stage of labor using instrumental delivery to reduce the need for excessive pushing.
If oxytocin is used to induce labor, it should be used with caution, as it may cause tachycardia and hypotension.
In this case, the patient had induced vaginal delivery. Because of a presumed diagnosis of preeclampsia, intravenous (IV) magnesium sulfate was used during labor to control her elevated BP and reduce the risk of seizure. IV magnesium was discontinued shortly after initiation due to a severe hypotensive reaction.
Once the diagnosis of PGL/PHEO is confirmed, either with hormonal testing or surgical pathology, genetic counseling should be offered even if the family history is negative or unknown. Approximately 30% to 40% of individuals with an isolated PGL/PHEO have an underlying genetic cause for this diagnosis.7 There is an even stronger indication for genetic testing when PGL/PHEO is diagnosed during pregnancy, given the relatively young age of patients. The susceptibility genes that have been associated with PGL/PHEO include NF1, RET, VHL, succinate dehydrogenase subunits (SDHA, SDHB, SDHC, and SDHD), co-factor for succinate dehydrogenase complex (SDHAF2), and MEN1.
This case illustrates the importance of a timely diagnosis of paraganglioma/pheochromocytoma during pregnancy, as it can dramatically affect maternal and fetal mortality rates. High clinical suspicion for these tumors as a cause of elevated BP during pregnancy is critical for timely diagnosis, particularly in patients who lack other characteristics typical of pregnancy-related hypertensive disorders (proteinuria, edema, etc.) or who present with an elevated blood pressure early in pregnancy.
1 Neumann HPH, Young Jr WF, Eng C. Pheochromo-cytoma and Paraganglioma. N Engl J Med. 2019; 381(6): 552–565.
2 Quartermaine G, Lambert K, Rees K, et al. Hormone-secreting Adrenal Tumours Cause Severe Hypertension and High Rates of Poor Pregnancy Outcome: A UK Obstetric Surveillance System Study With Case Control Comparisons. BJOG. 2018; 125: 719–27.
3 Schenker JG, Granat M. Pheochromocytoma and Pregnancy–An Updated Appraisal. Aust N Z J Obstet Gynaecol. 1982; 22: 1–10.
4 Donatini G, Kraimps JL, Caillard C, et al. Pheochromocytoma Diagnosed During Pregnancy: Lessons Learned From a Series of Ten Patients. Surg Endosc. 2018; 32: 3890–900.
5 van der Weerd K, van Noord C, Loeve M, et al. Endocrinology in Pregnancy: Pheochromocytoma in Pregnancy: Case Series and Review of Literature. Eur J Endocrinol. 2017; 177: R49–R58.
6 Wing LA, Conaglen JV, Meyer-Rochow GY. Paraganglioma in Pregnancy: A Case Series and Review of the Literature. J Clin Endocrinol Metab. 2015; 100: 3202–3209.
7 Lenders et al. Pheochromocytoma and Pregnancy: A Deceptive Connection. Eur J Endocrinol. 2012;
166: 143–150.
