


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
7 Minutes
Pouneh K. Fazeli, MD, MPH, and Anamil Khiyami, MD, were the authors of this case study.
A 23-year-old woman with no prior medical history presented to UPMC Presbyterian with a headache, nausea, and vomiting.
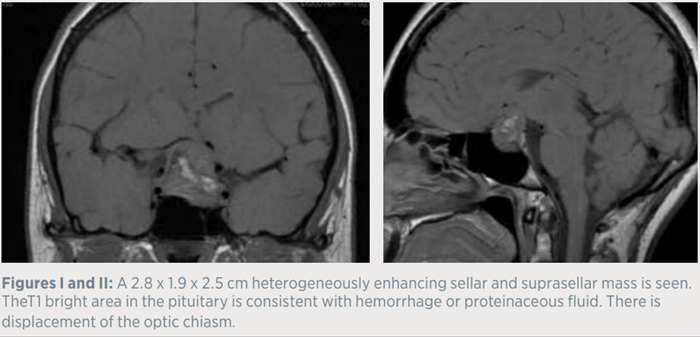
Three days prior to admission, the patient developed a sudden onset headache associated with nausea and vomiting. A brain MRI was performed in the emergency department and demonstrated a 2.8 cm sellar/suprasellar mass with evolving hemorrhage, suspicious for apoplexy. The neurosurgical and neuroendocrine teams were subsequently consulted. The patient reported regular monthly menstrual cycles and was not taking oral estrogen. She denied symptoms related to hyper or hypothyroidism. She also denied any changes in weight, and reported no history of nausea, vomiting, or orthostasis prior to this presentation. In addition, she was not experiencing any changes in shoe size or ring size, and denied galactorrhea, nocturia, polydipsia, or polyuria.
The patient was not taking any medications, vitamins, or supplements and denied using tobacco and EtOH. She reported working in a tire factory and following a vegan diet. The patient’s family history was remarkable for a maternal grandfather and maternal uncle with nephrolithiasis. There was no reported history of pituitary tumors or neuroendocrine tumors.
On examination, the patient was hemodynamically stable, and her BMI was low at 17.4 kg/m2. She did not appear Cushingoid or acromegalic. Her neurologic exam did not demonstrate any focal deficits, and ophthalmologic assessment was consistent with subjective blurring in her right peripheral visual field.
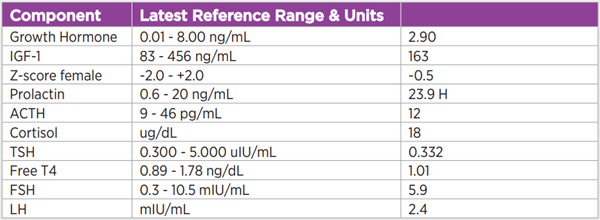
An evaluation was undertaken to assess for a functional adenoma as well as hypopituitarism, given the size of the sellar mass and the concern for apoplexy. Laboratory values were notable for a normal insulin-like-growth-factor 1 [IGF-1] level, normal TSH/Free T4, and a slightly elevated prolactin level (23.9 ng/mL), which was confirmed by serial dilution. A cortisol level of 4 mcg/dL was considered inappropriately low in the setting of acute illness, and stress-dose glucocorticoids were initiated. Given the clinical and radiologic findings suggestive of pituitary apoplexy, the patient underwent emergent endoscopic endonasal resection of her tumor. Surgical pathology demonstrated a pituitary adenoma which was sparsely granulated and stained for growth hormone (GH). An IGF-1 level measured six weeks postoperatively was normal.
As suggested by its name, the primary function of the GH/IGF-1 axis is to mediate growth. Importantly though, GH has another critical metabolic function. GH is a counter-regulatory hormone that allows us to survive prolonged periods of fasting; growth hormone has both lipolytic and insulin resistance effects.1 Therefore, in patients with chronic caloric deprivation, such as those with anorexia nervosa, normal or high levels of GH are an adaptive response to the state of starvation; minimizing energy expenditure on growth is also an adaptive response, and IGF-1 levels are low in states of starvation.2 This combination of adaptive responses is referred to as GH resistance.2 GH resistance allows for the exploitation of the beneficial counter-regulatory effects of GH during periods of starvation, while minimizing energy expenditure on growth.
On the other end of the spectrum, in states of appropriate caloric intake, GH promotes anabolism by stimulating IGF-1 secretion from the liver. Normal functioning of this axis is essential during childhood and puberty for skeletal maturation. Among other anabolic effects, IGF-1 promotes bone acquisition via promotion of osteoblast differentiation.3
Acromegaly, a disorder characterized by GH excess, is a rare and underdiagnosed disorder of the GH/IGF-1 axis. In well over 90% of cases, acromegaly results from GH hypersecretion from a pituitary adenoma. Much more rarely, it can be due to hypothalamic or ectopic GHRH hypersecretion or due to ectopic GH production. The GH hypersecretion leads to overproduction of IGF-1, which typically results in multiple clinical signs/ symptoms, including soft-tissue growth, hypertension, insulin resistance, and arthropathy.4 Patients suspected of having acromegaly are screened with an IGF-1 level. If this level is definitively elevated in the setting of signs/symptoms of acromegaly, no further testing is needed to make the clinical diagnosis of acromegaly. In patients whose diagnosis cannot be confirmed with an IGF-1 level alone, further dynamic testing with an oral glucose tolerance test should be performed. Individuals who fail to suppress GH to less than 1 ng/ml [or < 0.4 ng/mL depending on the sensitivity of the assay] after a glucose load are diagnosed with acromegaly.5
Silent somatotroph adenomas, defined as pituitary adenomas that stain for GH, but are not associated with biochemical evidence of GH excess, account for approximately 2-4.2% of pituitary adenomas.6–8 In contrast, some individuals will have biochemical evidence of acromegaly without clinical features, and this has been termed “clinically silent acromegaly.”9
In 2017, Langlois et al. compared silent somatotroph adenomas to silent gonadotroph adenomas, which is the most frequent type of nonfunctional adenoma, and found that silent somatotroph adenomas occurred more frequently in women, presented at an earlier age, and were more likely to recur. There was a 29% recurrence rate after a mean follow-up of approximately four years.7
As diagnosis of clinically evident acromegaly is often delayed many years, one hypothesis as to why silent somatotroph adenomas are biochemically and clinically silent is that acromegaly is a spectrum with silent somatotroph adenomas representing the first phase of the disease, and “clinically silent acromegaly” representing another phase early in the disease course before physical changes have occurred. The transition from silent somatotroph adenomas to more clinically apparent disease has been reported by Langlois et al.7 Two patients with silent somatotroph adenomas in their series developed an elevated IGF-1 level on a follow-up, including a 38-year-old woman whose IGF-1 level increased to 1.5 times the upper limit of normal, and who developed symptoms of arthralgias in the setting of the IGF-1 elevation.7 Other proposed theories for the lack of biochemically evident GH excess include the production of a biologically inactive hormone.10
There are several other factors that can affect IGF-1 levels, one of which is estrogen. Oral estrogen has been shown to suppresses IGF-1.11 Estrogen lower IGF-1 levels, in part by increasing GH binding protein concentrations, and also through disruption of GH signaling.12 Therefore, estrogen may decrease IGF-1 levels into the normal range, especially in early acromegaly.
Another factor that can affect IGF-1 levels is nutritional status. GH resistance is an adaptive response to chronic undernutrition. In a state of GH resistance, levels of GH (a lipolytic hormone with insulin-resistant effects) are normal or elevated, but IGF-1 levels (which mediate most of the growth-related actions of GH) are low. These levels minimize energy expenditure on growth. By observing the effects of a 10 day fast on the GH/IGF-1 axis, Clemmons et al. demonstrated the development of GH resistance during acute starvation. A sharp drop in IGF-1 levels to below normal range within five days of fasting, concomitant with an eventual increase in GH levels, was observed during the 10 day fast.13
Anorexia nervosa, a psychiatric disorder characterized by self-imposed chronic starvation and an inability to maintain a normal BMI, is a human model of chronic starvation. Anorexia nervosa is a state of GH resistance, and adolescent girls with anorexia nervosa have significantly higher nadir GH levels in response to an oral glucose load compared to normal-weight controls.14 Fazeli et al. performed a randomized study of rhGH in individuals with anorexia nervosa.15 Despite supraphysiologic doses of rhGH, levels of IGF-1 were similar in both the rhGH and placebo groups, suggesting that GH resistance cannot be easily overcome in states of undernutrition. Similarly, isolated protein deficiency also results in GH resistance,16 as do some isolated vitamin deficiencies.2
This patient had numerous possible reasons to account for a normal IGF-1 level preoperatively. Her BMI was low (in the anorexia nervosa range), and she followed a vegan (low protein) diet, both of which could lead to a state of GH resistance. A silent somatotroph adenoma also cannot be excluded in this case. Given her low BMI and poor protein intake, an oral glucose tolerance test was not performed due to the possibility that GH may not suppress in response to a glucose load in this setting.14 Therefore, the patient’s clinical, biochemical, and radiologic parameters will continue to be carefully and closely monitored.
Please refer to the Update in Endocrinology Spring 2022 CME course for a full list of references featured in this article.
