


Password Reset
Forgot your password? Enter the email address you used to create your account to initiate a password reset.


Forgot your password? Enter the email address you used to create your account to initiate a password reset.
5 Minutes
By Quyen Nguyen, MD, and Michael G. Risbano, MD, MA, FCCP
Case Introduction
A 60-year-old WHO functional Class II female with a past medical history of hypothyroidism, sclerosis sine scleroderma (ANA 1:640 nucleolar, elevated aldolase levels and antibodies to Th/To), nonspecific interstitial pneumonia (NSIP), and pulmonary arterial hypertension, followed up at the UPMC Comprehensive Pulmonary Hypertension Clinic for ongoing dyspnea on exertion. She noted progressive dyspnea on exertion over the previous year, in particular on hills and stairs. She was treated with an upfront combination therapy of tadalafil and ambrisentan.1,2
In addition to pulmonary vasodilators, she was on aspirin and levothyroxine. She had no known history of venous thromboembolism, diet drug use, amphetamine or amphetamine derivative use, illicit drug use, HIV, miscarriage, or blood transfusion. Her social history was significant for never smoking, occasional alcohol use, and no significant occupational exposures in her work as an attorney.
Physical exam: BP 147/98, P 116, oxygen saturation of 96% on room air. She appeared younger than her stated age. No jugular venous distension. Chest exam with normal air movement, no crackles or wheezing. Cardiac exam with an enhanced second pulmonary closure was sound. No pedal edema. Her skin exam revealed dactylitis with chapped finger tips, without skin thickening, telangiectasias, or ulcerations.
Pulmonary function testing: FVC 1.78 liters (56% predicted), FEV1 1.42 liters (64% predicted), FEV1/FVC ratio 80%, TLC 4.90L (77% predicted), and DLCO 22.91 (32% predicted).
Computed tomography angiography of the chest: Increase in subpleural interstitial markings and septal thickening consistent with nonspecific interstitial pneumonia (NSIP) pattern of interstitial lung disease (ILD) without honeycombing or fibrosis. There was no evidence of acute or chronic pulmonary embolus.
Echocardiogram: Normal left ventricular ejection fraction, mildly dilated right ventricle with TAPSE 1.8 cm, tricuspid regurgitation peak velocity of 3.3 m/s, and estimated pulmonary artery systolic pressure of 47 mmHg.
Ventilation/perfusion scan: Revealed no chronic thromboembolism.
Case Discussion and Initial Management
After one year on dual pulmonary vasodilator therapy, the patient had subjective improvement albeit persistent dyspnea on exertion, the etiology of which was unclear. She had mild radiographic progression of ILD, and echo showed decreased PASP to 22 mmHg.
Her six-minute walk distance was 365 m, where 300-400 m is considered intermediate risk for PAH.3,4 She was referred for invasive cardiopulmonary resistance to quantify her respiratory and cardiovascular limitation and determine the role for augmentation of pulmonary vasodilator therapy versus possible immunosuppression for her ILD and scleroderma.
Advanced Cardiopulmonary Exercise Testing Procedure description: The patient arrived at the cardiac catheterization lab at UPMC Presbyterian, was placed in the supine position, and was prepped and draped in a sterile fashion. A left radial arterial line and a right internal jugular 8F introducer was placed under ultrasound guidance. A Swan-Ganz VIP catheter was inserted. For the exercise portion, an ACPET was performed with the MGC Diagnostics metabolic cart. Continuous oxygen saturation, EKG, hemodynamics, and expired gas analysis were performed. Serial blood pressures were obtained from the arterial line. Resting seated and exercise measurements were obtained.
ACPET Discussion
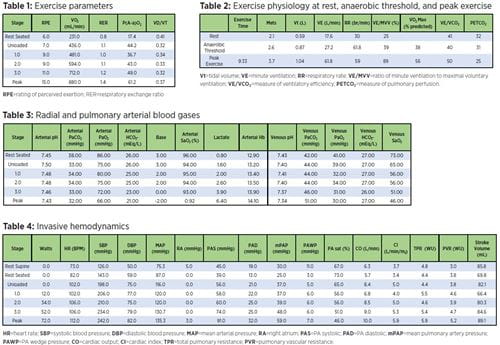
The patient performed a maximal exercise study as indicated by an RER >1.1. She achieved a low VO2 at anaerobic threshold and peak exercise indicating limitation of exercise capacity. There was little increase in VO2 as the work rate increased. The patient had multiple abnormalities present in this study. She had resting pulmonary arterial hypertension. With exercise, her pulmonary artery pressures and total and pulmonary vascular resistance increased. However, this may not be limiting her exercise capacity as her cardiac output increased to 88% predicted peak cardiac output (based upon her VO2 max achieved) at peak exercise. This indicates that it is unlikely that her pulmonary vascular disease was a limitation to exercise. Despite a maximal exercise result, the patient did not maximally extract oxygen at peak exercise as the pulmonary artery oxygen saturation was only 46%. At peak exercise this value can be as low as 25% in normal individuals. The lack of normal peripheral oxygen extraction at peak can be seen in patients with mitochondrial myopathies, which also results in a relatively elevated cardiac output at peak exercise despite low VO2 max. Some of the CO that should be low because of pulmonary vascular disease may be offset by the presence of a concomitant metabolic myopathy. The primary exercise limitation is mostly due to ventilatory limitation. At peak exercise, the patient had an elevated breathing frequency, low breathing reserve, elevated VD/VT, high VE/VCO2 at anaerobic threshold, and elevated A-a gradient.
References
1. Galie N et al. “Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension.” N Engl J Med. 2015. 373(9):834-44.
2. Coghlan JG et al. “Initial combination therapy with ambrisentan and tadalafil in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH): subgroup analysis from the AMBITION trial.” Ann Rheum Dis. 2017. 76(7):1219-1227.
3. McLaughlin VV, McGoon MD. “Pulmonary arterial hypertension.” Circulation. 2006. 114(13):1417-31.
4. Galie N et al. “Risk stratification and medical therapy of pulmonary arterial hypertension.” Eur Respir J. 2018.
